
钠离子电池(SIBs)具有成本低、安全性高、储量丰富等多重优点,在大规模储能领域备受世界瞩目。钠离子电池正极材料容量相对较低,如 Na0.78Ni0.23Mn0.69O2、NaVPO4F/C和 Na2CoFe(CN)6,限制了能量密度的提高。使用硫阴极的钠硫(Na-S)电池理论比容量(1,672 mAh g-1)和能量密度(1,274 Wh kg-1)较高,而且钠和硫的供应充足,因此被认为是一种很有前景的电池技术。尽管具有这些优势,Na-S 电池仍然面临许多挑战,包括 a) 硫的固有绝缘特性导致电荷转移差(~5.1*10-30 S cm-1),利用率低;b)硫具有较大的体积膨胀性(从 S8 到 Na2S 为 260%),无法实现稳定的容量输出;c)硫与钠氧化还原反应的中间产物主要是可溶性多硫化钠(Na2Sx,4 ≤ x ≤ 6),造成严重的穿梭效应;d)固态硫化钠的相关反应动力学迟缓。有必要制定有效的策略来应对上述挑战,以促进 Na-S 电池的实际应用。
碳材料具有良好的导电性、化学稳定性和可调性,在硫阴极方面具有巨大潜力。人们开发了碳材料的孔隙结构来承载硫。人们合成了一种由碳壳包裹的多腔碳纳米球组成的复杂微孔碳材料,从而实现了高硫负载,并为抑制多硫化物溶解提供了物理屏障。此外,通过调节微孔尺寸,可选择性地去除长链硫分子(Sx,x > 4),而保留合适的短链硫分子(Sx,x ≤ 4),这在一定程度上避免了中间多硫化物产物的不可逆溶解。例如,通过热解聚偏二氟乙烯(PVDF)制备的超微孔碳(0.55 nm)能够容纳 S2 ˉ S4 小分子。不过,这种将小分子硫限制在超微孔中的方法对碳基质的设计要求很高,无法从根本上解决多硫化物的形成和溶解问题。
共价化学键表现出硫对保护硫阴极的强大锚定能力。通过共价键将硫接枝到碳材料上的化学方法是抑制多硫化物穿梭的有效方法。据报道,将苯并二噻吩-4,8-二酮和硫粉混合并热解,可构建共价硫基碳质材料。虽然在一定程度上避免了 Na2Sx(4 ≤ x ≤ 8)的形成,但其循环性能仍然较差,循环 50 次后容量保持率仅为 61.6%。以苯基膦酸为碳源/催化剂,硫酸钠为硫前驱体/盐模板,合成了共价硫碳桥杂化物。在放电过程中,C-S 和 S-S 键断裂形成 Na2S,碳骨架发生不可逆的异构化,导致 200 次循环后容量保持率较低,仅为 78.3%。硫也可以通过高温蒸发渗透法共价锚定在多巴胺涂层的氮掺杂二维碳石墨烯基底上。在放电过程中,短链硫与 Na+ 发生反应,C-S 键断裂形成 Na2S,由于硫的严重流失,硫的利用率很低,仅为 75.6%(第二周期)。上述硫的主要来源是无机硫粉。有机硫化合物因其固有的含 S 共价键可避免穿梭效应而受到广泛关注。研究发现,4,4′-硫代二苯硫酚作为一种有机硫阴极材料,在充电过程中会形成由两个(S-R-S)n 组成的有机硫低聚物,从而确保了良好的稳定性和 210 mAh g-1 的可逆容量。然而,有机硫分子中有限的硫含量似乎阻碍了比容量的提高。有机多硫化物由于含有多种硫,可能会获得较高的比容量。据报道,与二甲基二硫相比,二甲基三硫可提供较大的比容量,因为额外的中间硫原子可提高比容量。由二吡啶基二硫化物和硫合成的一系列二吡啶基多硫化物(如 Py2S3、Py2S4、Py2S5、Py2S6、Py2S7 和 Py2S8)显示出可逆的电化学行为。然而,有机硫化合物的导电性较低,需要过度使用导电添加剂,这无疑会降低电池的整体能量密度。一般来说,引入共价键硫是解决硫阴极稳定性问题的一种好方法,但仍会遇到容量衰减和/或导电率低的挑战。此外,由于 C-S 键比 C-C/C=C 键更长,键能更低,需要调节电子和化学环境,因此从无机硫粉中构建共价 C-S 键也面临着原子层面的挑战。
【工作要点】
在本研究中,作者报告了一种化学和空间双封闭工程,从根本上解决了稳定 Na-S 电池的硫相关问题。通过热切割大分子 S8 生成小硫组分(S1 和 S2),然后密封在碳材料的封闭孔隙中,同时通过 N 原子调节电子和化学环境构建共价 C-S/N-C=S 键。作者利用原位傅里叶变换红外图谱(FTIR)对双封闭硫材料的构造进行了探测。利用飞行时间二次离子质谱(TOF-SIMS)检测了碳材料中硫的存在形式。系统评估了硫阴极的电化学性能,如比容量和循环稳定性。通过紫外-可见(UV-vis)吸收图谱、X 射线衍射(XRD)、X 射线光电子能谱(XPS)和理论计算等多种图谱分析,深入研究了钠储存过程中 C-S 共价键的演化机理。
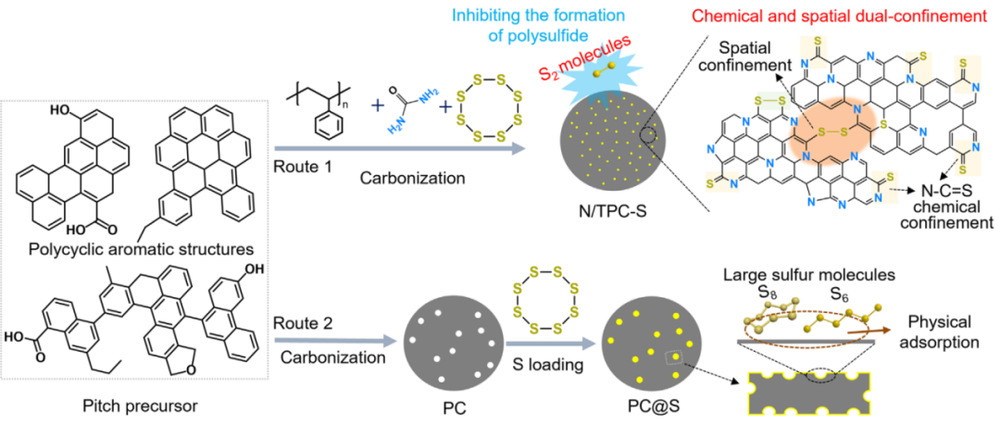
图 1.硫碳复合材料合成工艺的比较(工艺 1:空间和化学双重强化法;工艺 2:传统熔融硫注入法)
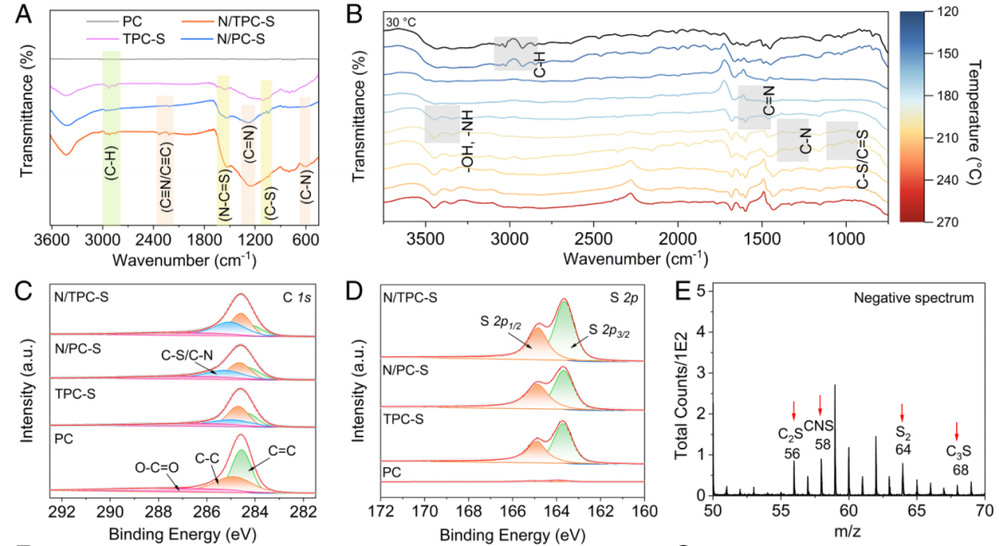
图 2.PC、TPC-S、N/PC-S 和 N/TPC-S 的结构特征。(a) 傅立叶变换红外图谱。(b) 通过原位傅立叶变换红外图谱研究了在 PS 微球、尿素和硫的存在下热处理沥青过程中化学键的演变。(c) C 1s XPS 图谱。(d) S 2p XPS 图谱。(E-G) N/TPC-S 的 TOF-SIMS 数据。
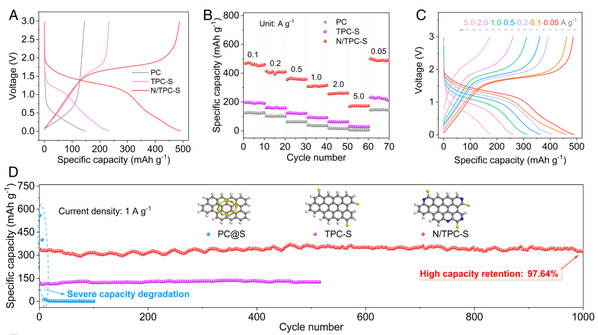
图 3.PC、TPC-S 和 N/TPC-S 的电化学性能。(a) 0.05 A g-1 时的 GCD 曲线。(B)速率性能。(c) N/TPC-S 在不同电流密度下的 GCD 曲线。(d) 1 A g-1 时的循环性能。(e) 5 A g-1 时 N/TPC-S 的循环性能和相应的库仑效率。
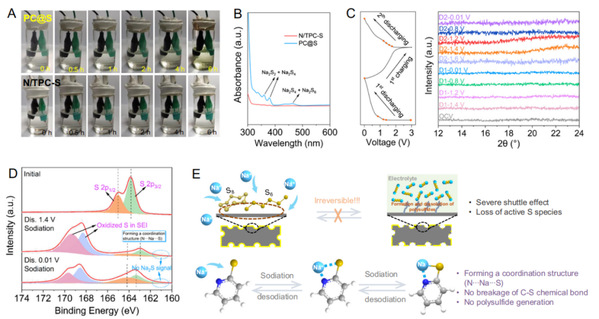
图 4. (a) PC@S 和 N/TPC-S 正极的小瓶电池在初始状态和放电 0.5、1、2、4 和 6 小时后的照片。(d) N/TPC-S 在不同充放电状态下的高分辨率 S 2p XPS 图谱。(e) PC@S 和 N/TPC-S 储钠机理示意图(灰色球:C 原子,深蓝色球:N 原子):C原子,深蓝色球N原子,黄色球S 原子,亮蓝球:钠离子)。
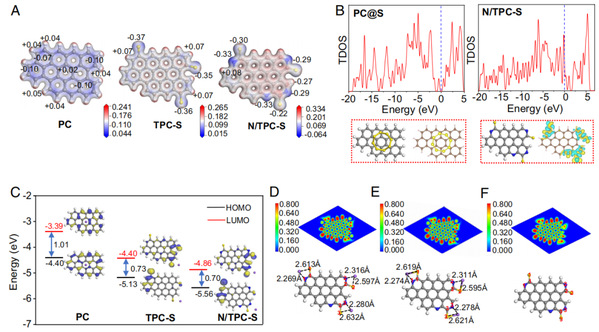
图 5. (a) PC、TPC-S 和 N/TPC-S 的静电势。(b) PC@S 和 N/TPC-S 的 TDOS 和优化结构以及相应的 DCD 图(等表面水平 = 0.005)。(c) PC、TPC-S 和 N/TPC-S 结构的电子密度以及 LUMO 和 HOMO 的能量(eV)。(d) 放电初期、(e) 放电后期和 (F) 充电过程中 N/TPC-S 的 ELF 分析和优化配置。
【结论】
作者报告了一种化学和空间双掺杂法合成先进 S 阴极(N/TPC-S)的方法,并研究了 Na-S 电池的性能和钠存储机制。通过对多种图谱分析的系统研究发现,在 N 原子的帮助下,S 与碳原子共价键合(C-S/N-C=S 键),调节了碳基体的电子和化学环境。此外,S8 大分子转化为 S1-S2 小分子,封存在碳材料的密闭孔隙中。因此,N/TPC-S 阴极表现出良好的电化学性能,在 1000 次循环后仍能保持约 100% 的高容量,优于目前 Na-S 电池的 S 阴极。紫外可见光、原位 X 射线衍射、原位 XPS 和光学观察结果表明,可溶性多硫化物和 Na2S 均未被破坏,因此具有良好的循环稳定性。系统的理论计算表明,钠离子在放电过程中不会断开 C-S 键,而是在 N 原子和 S 原子之间的位点形成配位结构(N--Na--S),因此在反复充放电过程中不会形成 Na2S。化学和空间限制工程为锁定活性 S 成分提供了一种有效的方法,从而解决了 Na-S 电池和其他 S 基电池与 S 有关的难题。
该论文的第一通讯单位为北京化工大学,第一作者是北京化工大学化学工程学院博士生张永,通讯作者是北京化工大学化工学院,化工资源有效利用国家重点实验室邱介山教授、杨琪教授和太原理工大学章日光教授。
原文链接:
https://www.pnas.org/doi/10.1073/pnas.2314408120